 |
# Construct the appropriate matrices/data frames for
# construction of the heat map.
ii.mat <- exprs.eset[ii,]
ii.df <- data.frame(ii.mat)
# Dislplay ii.df
ii.df
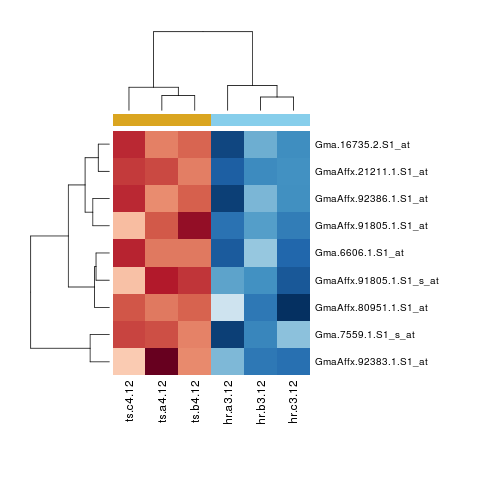
# Construct heatmap using reds and blues
library('RColorBrewer')
library('genefilter')
hmcol <- colorRampPalette(brewer.pal(10, 'RdBu'))(256)
tv.Hawaii.Resistant <- dimnames(ii.mat)[[2]][1:3]
spcol <- ifelse(dimnames(ii.mat)[[2]] == tv.Hawaii.Resistant, 'skyblue', 'goldenrod')
heatmap(ii.mat, col = hmcol, ColSideColors = spcol, margins = c(10,15))
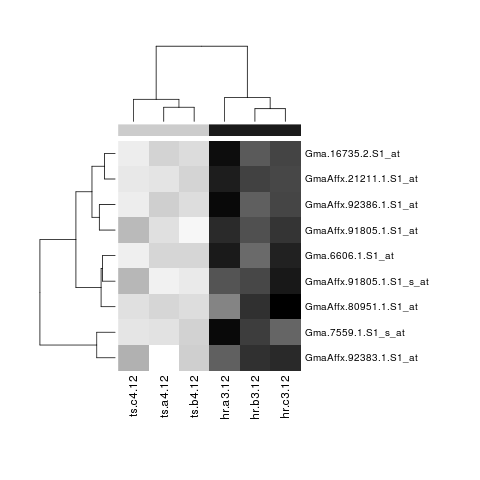
# Construct heatmap using a grey (gray) scale
library('RColorBrewer')
library('genefilter')
hmcol <- colorRampPalette(brewer.pal(9, 'Greys'))(256)
tv.Hawaii.Resistant <- dimnames(ii.mat)[[2]][1:3]
spcol <- ifelse(dimnames(ii.mat)[[2]] == tv.Hawaii.Resistant, 'grey10', 'grey80')
heatmap(ii.mat, col = hmcol, ColSideColors = spcol, margins = c(10,15))
(Complete File)(Rout)
|
{kind=link}
{kind=link}